

About our research
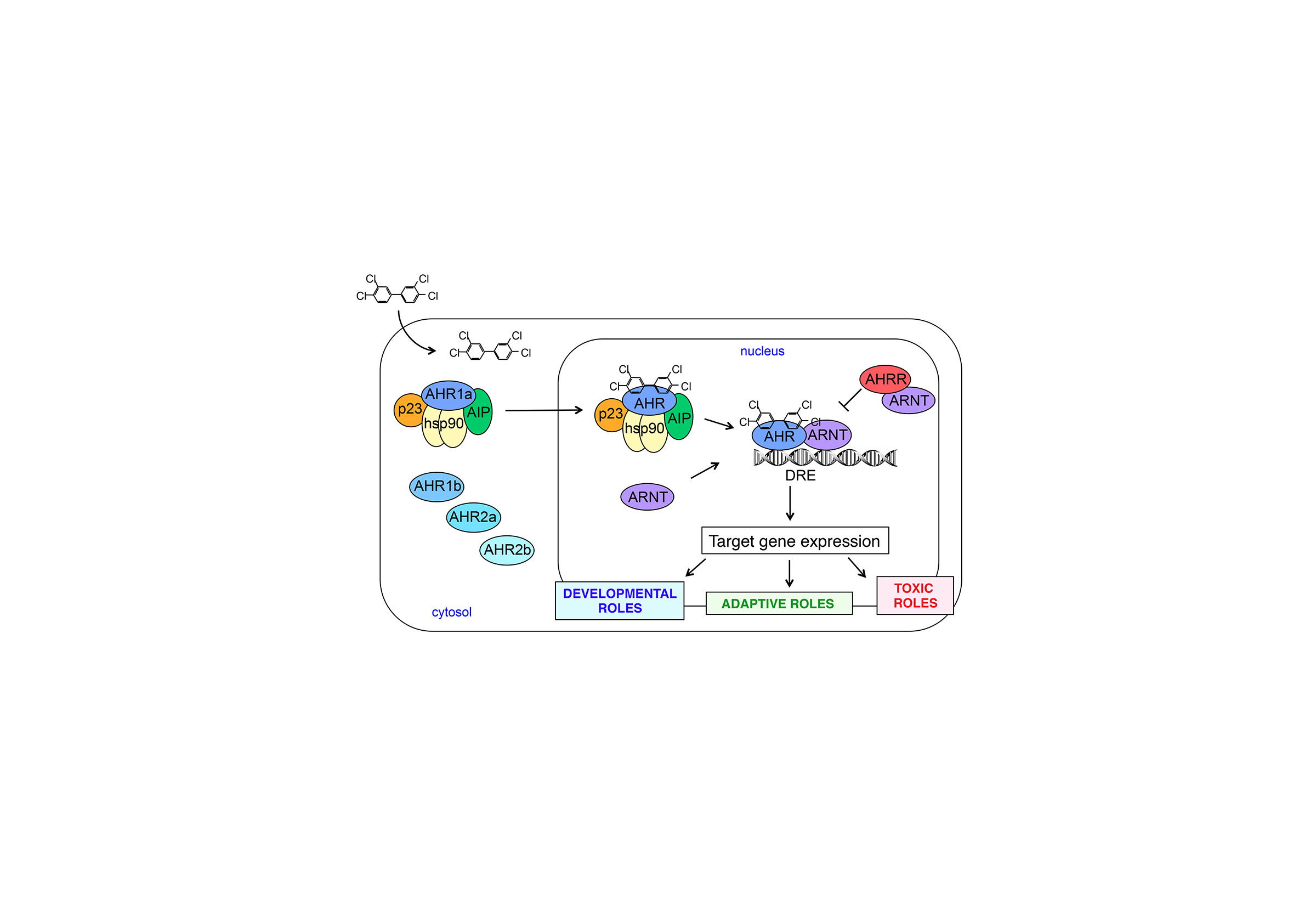
The overall objective of research in our laboratory is to understand the biochemical and molecular mechanisms that underlie the interactions of animals with their chemical environment. We examine these mechanisms from comparative and evolutionary perspectives in order to understand the fundamental features of the biochemical systems that control the response of animals to toxic chemicals. We conduct research on a variety of animal species (fish, birds, whales) as well as using established biomedical model systems such as the zebrafish embryo and human cells in culture.
Our research is guided by general questions such as:
- How did chemical signaling pathways evolve in metazoans?
- What is the role of these pathways in adaptation to long-term chemical exposure?
- What is the mechanistic basis for differential sensitivity to chemicals among species and populations of animals?
- What are the mechanisms by which chemicals disrupt embryonic development?